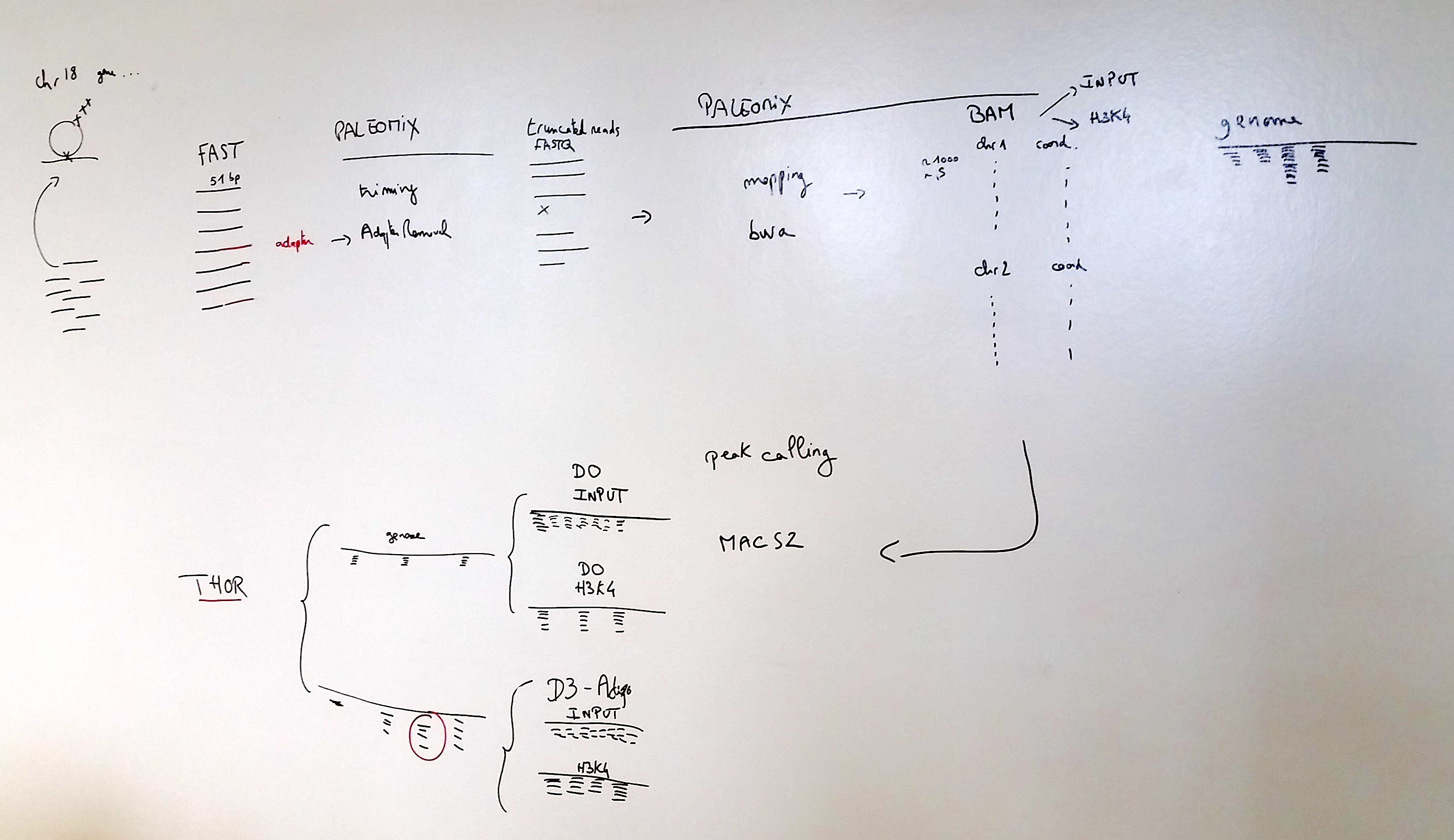
Workflow
The workflow of all steps is summarised below:
load the singularity container
Singularity allows to use containers (from i.e Docker) on High-Performance Computer. For more details see the lecture by Valentin Plugaru
Shortly, we built a container with all the necessary tools and softwares embeded. Hence, you just need to book the HPC resources and load the container to start working on your chip-seq sequences.
Tweak for the picard
we need to tweak this location only once.
mkdir -p ~/install/jar_root/
cp /scratch/users/aginolhac/picard.jar ~/install/jar_root/
book resources on iris
- 2 hours
- 8 cores
- interactive
srun --cpu-bind=none -p interactive --time=2:0:0 -c 8 --pty bash -i
load the container
- first we load the tools
singularity - second we load the container
module load tools/Singularity
singularity shell -s /bin/bash --bind /scratch/users:/scratch/users /scratch/users/aginolhac/ubuntu-chip-seq.simg
then you are inside the container, all the tools should be available.
try running the following and raise your hand if any is command not found
bwa
samtools
macs2
paleomix
prepare your working environment
- go to your home directory:
cd - create a new folder to work in:
mkdir chip-seq - go inside:
cd chip-seq - create and go in a sub-folder:
mkdir fastq - go inside:
cd fastq - symbolic link the FASTQ files:
ln -s /scratch/users/aginolhac/chip-seq/fastq/C* . - check your actions:
ll(alias ofls -l)
check integrity of files
Just as a side note, such large files are usually a pain to download. Since they are the very raw files
after the sequencer (despite basecalling) checking their integrity is worth doing.
Computing the md5sum ensure you have the same file as your sequence provider.
Then paleomix will check the FASTQ are correct, i. e have 4 lines in a correct format.
md5sum -c C53CYACXX_TC1-I-A-D3_14s006682-1-1_Sinkkonen_lane114s006682_sequence.txt.md5
you should observe an OK after few seconds of computing time.